

买房后要担心的可远不止甲醛,地板上的问题更是让人防不胜防
来源: 发布时间:2021-01-22
装修问题如今已经是社会大众及其敏感的一个重要方面,城市的生存空间逐渐狭窄,因而室内装修的安全与质量问题必然会遭到社会的监督,装修材料更要严格。近来,针对消费者投诉、举报集中或以往抽检发现质量问题较多的产品,上海市市场监管局集中执法力量对宝山、嘉定2个区域内19家企业和天猫商城、东方CJ 2个网络交易平台销售的64批次人造板、地板进行了监督抽查。
经检测,有12批次不合格,不合格检出率为18.8%,其中,抽查人造板39批次,有9批次不合格,抽查地板25批次,有3批次不合格。人造板的不合格项目为甲醛释放量、表面耐香烟灼烧;地板的不合格项目为静曲强度、面板木材名称、标志或包装标签(标记)。

人造板是装修材料中非常常见的一种材料,目前我国已成为世界人造板等室内装饰装修材料的生产大国和消费大国。2011年以来,我国人造板产量居世界第一,2015年,人造板产量已达2.87亿m3。而甲醛释放限量是人造板及其制品的重要环保性能指标。
人造板中藏着的隐患
人造板中的甲醛存在于生产的各个环节,几乎让人防不胜防。
首先人造板中胶粘剂自身含有游离的甲醛。生产人造板时使用的胶黏剂脲醛树脂在一定条件下分离出甲醛。其次,生产人造板使用原料也会影响甲醛的产生。木材的穿孔值为1~3mg/100g,木材在干燥时会部分分解,生成醋酸与蚁酸,木素中甲氧基断链从而释放出甲醛。
同时,人造板制板工艺也会对甲醛的产生造成影响。板材芯层甲醛散发潜势较高,原因是芯层为固体“薄弱区”,其温度较低、含水率较高、PH值也会较低、固化程度差,容易水解而生产甲醛。结构降解释也会放出甲醛。温度、湿度、酸碱、风化、光照等环境使板内未完全固化的树脂发生降解而释放出甲醛。
此次抽检出某品牌人造板甲醛释放量实测为0.330mg/m³,国家标准GB18580-2017《室内装饰装修材料人造板及其制品中甲醛释放限量》规定甲醛释放限量值为0.124mg/m3,限量标识为E1,远超限量值。但是,甲醛达到一定浓度后,会对人体产生一定的危害。
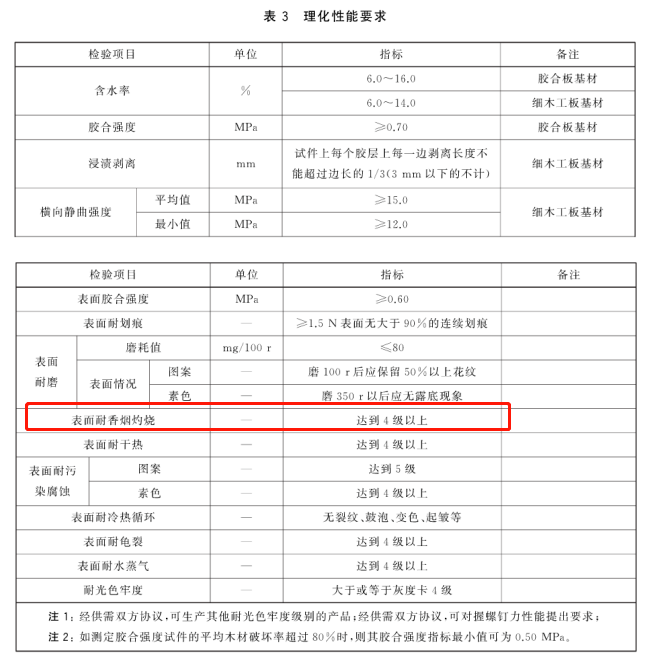
人造板在装修中应用广泛,其分类也繁多。较为主流的有细木工板、装饰单板贴面人造板、浸渍胶膜纸饰面胶合板、浸渍胶膜纸饰面细木工板等。其中在此次抽检中,某品牌浸渍胶膜纸饰面细木工板以及某品牌的浸渍胶膜纸饰面胶合板就出现了表面耐香烟灼烧测试出现明显的棕色斑,表面耐香烟灼烧等级实测为2级。
依据GB/T 34722-2017 《浸渍胶膜纸饰面胶合板和细木工板》,要求应达到4级以上,与标准要求不符。表面耐香烟灼烧是评价材料表面装饰层对点燃香烟灼烧的抵抗能力,该指标不合格,影响产品的使用寿命和使用效果。
地板
地板是装修中必不可少的,因而成为了此次抽检的重点项目。地板的分类较多,市面上较为常见有仿古浸渍纸层压木质地板、浸渍纸层压木质地板、实木地板等。此次抽检为某品牌的浸渍纸层压木质地板,静曲强度实测为平均值为22.6MPa。
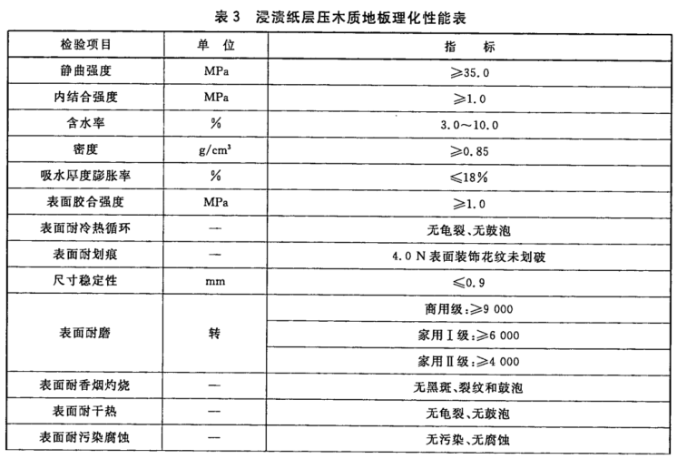
而根据GB/T 18102-2007《浸渍纸层压木质地板》规定,静曲强度应为MPa≥35 MPa,因此不合格。静曲强度不合格,影响地板的支撑力度和铺装后的平稳性。
同时,此次还抽检出实测面板木材名称与企业标称不符、企业未标示与产品相匹配的标准等问题。但是标志或包装标签是消费者了解产品信息的重要渠道,若面板木材名称、标志或包装标签不合格,容易误导消费。
现代社会,人们在室内的生存时间逐渐延长,但生存空间却在不断压缩,这就导致人们对装修材料的每一项决定都慎重其事,人造板、地板产品质量也成为了社会关注的热点,尤其是甲醛释放量超标等焦点问题,一直是消费者所重视的。因而人造板以及地板相关企业必然要严肃面对如今的社会新形势,加强对自身的检测,才能正确应对市场监管,避免对企业社会声誉以及未来可持续发展造成严重危害。

-

-
 400-182-9001
400-182-9001 -


